Kinetic Monte Carlo
Kinetic Monte Carlo (KMC) algorithms are composed of various methods for stochastically simulating chemically reacting systems.
While various flavors of KMC are encountered in different fields of science, KMC in the context of chombo-discharge is primarily associated with chemistry kernels.
Concept
In chombo-discharge the Kinetic Monte Carlo solver advances a state (or multiple states) represented e.g. as state vectors
Each row in \(\vec{X}\) represents the population of some chemical species, and is indexed by an integer. Reactions between species are represented stoichiometrically as
where \(k\) is the reaction rate. Each such reaction is associated with a state change in \(\vec{X}\), e.g.
where \(r\) is the reaction type and \(\nu_r\) is the state change associated with the firing of one reaction of type \(r\). A set of such reactions is called the reaction network \(\vec{R}\).
Propensities \(a_r\left(\vec{X}\right)\) are defined such that \(a_r\left(\vec{X}\right)\textrm{d}t\) is the probability that exactly one reaction of type \(r\) occurs in the time interval \([t, t+\textrm{d}t]\). For unimolecular reactions of the type
with \(A \neq B \neq \ldots\) the propensity function is \(k X_A X_B \ldots\). For bimolecular of the type
the propensity is \(k \frac{1}{2} X_A(X_A-1)\) because there are \(\frac{1}{2}X_A(X_A-1)\) unique pairs of molecules of type A. Propensities for higher-order reactions can then be expanded using the binomial theorem. For example, for a third-order reaction \(3X_A\xrightarrow{k} \emptyset\) the propensity function is \(k\frac{1}{6}X_A(X_A-1)(X_A-2)\).
Various algorithms can be used for advancing the state \(\vec{X}\) for an arbitrary reaction network \(\vec{R}\).
The Stochastic simulation algorithm (SSA). The SSA is also known as the Gillespie algorithm [Gillespie, 1977], and is an exact stochastic solution to the above problem. However, it becomes inefficient as the number of reactions per unit time grows.
Tau leaping, which is an approximation to the SSA which uses Poisson sampling of the underlying reactions.
Hybrid advance that switches between the SSA and tau-leaping in their respective limits, see Hybrid algorithm. The hybrid algorithm is taken from Cao et al. [2006], and switches between tau leaping and the SSA in their respective limits.
Stochastic simulation algorithm
For the SSA we compute the time until the next reaction by
where \(A = \sum_{r\in\vec{R}} a_r\) and \(u_1\) is a uniformly distributed random variable between \(0\) and \(1\). The type of reaction that fires is determined from
where and \(u_2\) is another uniformly distributed random variable between \(0\) and \(1\). The state is then advanced as
Tau leaping
With tau-leaping the state is advanced over a time \(\Delta t\) as
where \(\mathcal{P}\) is a Poisson-distributed random variable. Note that tau leaping may fail to give a thermodynamically valid state, and should thus be used in combination with step rejection.
Tau-leaping variants
The following forms of tau-leaping are also supported:
Midpoint tau-leaping.
Poisson random-corrections tau-leaping.
Implicit Euler-type tau-leaping.
These methods can be used either as standalone methods or together with the hybrid algorithm.
Warning
We do not recommend implicit methods for reactive problems. The reason for this is that exponential growth follows the equation
where \(k>0\) is a growth rate. Application of the implicit Euler rule to this system yields
which has a pole at \(\Delta t = k^{-1}\), and which is only non-negative for \(k\Delta t < 1\). Similarly, the discretization can then lead to a large overshoot. In practice, the time step then has to be limited to \(\Delta t < k^{-1}\).
On the other hand, an explicit Euler update would yield
which is stable for any \(\Delta t\) (provided \(k\) is positive).
Hybrid algorithm
The hybrid algorithm is taken from Cao et al. [2006]. Assume that we wish to integrate over some time \(\Delta t\), which proceeds as follows:
Let \(\tau = 0\) be the simulated time within \(\Delta t\).
Partition the reaction set \(\vec{R}\) into critical and non-critical reactions. The critical reactions are defined as the subset of \(\vec{R}\) that are within \(N_{\textrm{crit}}\) firings away from exhausting one of its reactants. The non-critical reactions are defined as the remaining subset.
Compute time steps until the firing of the next critical reaction, and a time step such that the propensities of the non-critical reactions do not change by more than some relative factor \(\epsilon\). Let these time steps be given by \(\Delta \tau_{\textrm{c}}\)and \(\Delta \tau_{\textrm{nc}}\).
Select a reactive substep within \(\Delta t\) from
\[\Delta \tau = \min\left[\Delta t - \tau, \min\left(\Delta \tau_{\textrm{c}}, \Delta \tau_{\textrm{nc}}\right)\right]\]Resolve reactions as follows:
If \(\Delta \tau_{\textrm{c}} < \Delta \tau_{\textrm{nc}}\) and \(\Delta \tau_{\textrm{c}} < \Delta t - \tau\) then one critical reaction fires. Determine the reaction type using the SSA algorithm.
Next, advance the state using tau leaping for the non-critical reaction.
Otherwise: No crical reactions fire. Advance the state using tau-leapnig for the non-critical reactions only. An exception is made if \(A\Delta\tau\) is smaller than some specified threshold in which case we switch to SSA advancement (which is more efficient in this limit).
Check if \(\vec{X}\) is a thermodynamically valid state.
If the state is valid, accept it and let \(\tau \rightarrow \tau + \Delta\tau\).
If the state is invalid, reject the advancement. Let \(\Delta\tau_{\textrm{nc}} \rightarrow \Delta \tau_{\textrm{nc}}/2\) and return to step 4).
If \(\tau < \Delta t\), return to step 2.
The Cao et al. [2006] algorithm requires algorithmic specifications as follows:
The factor \(\epsilon\) which determines the non-critical time step.
The factor \(N_{\textrm{crit}}\) which determines which reactions are critical or not.
Factors for determining when and how to switch to the SSA-based algorithm in step 5b.
Implementation
In chombo-discharge, the KMC solver is implemented as
/**
@brief Class for running Kinetic Monte-Carlo simulations.
@details The template parameter State is the underlying state type that KMC operators on. There are rather simple
required member functions on the State parameter, but the reaction type (template parameter R) MUST be able to
operate on the state through the following functions:
1. Real R::propensity(State) const -> Computes the reaction propensity for the input state.
2. T R::computeCriticalNumberOfReactions(State) const -> Computes the minimum number of reactions that exhausts one of the reactants.
3. void R::advanceState(State&, const T numReactions) const -> Advance state by numReactions.
4. std::<some_container> getReactants() const -> Get reactants involved in the reactions.
5. static T R::population(const <some_type> reactant, const State& a_state) -> Get the population of the input reactant in the input state.
The template parameter T should agree across both R, State, and KMCSolver. Note that this must be a signed integer type.
*/
template <typename R, typename State, typename T = long long>
class KMCSolver
{
public:
/**
@brief Alias for the list of reactions.
*/
using ReactionList = std::vector<std::shared_ptr<const R>>;
/**
@brief Full constructor.
@param[in] a_reactions List of reactions.
*/
inline KMCSolver(const ReactionList& a_reactions) noexcept;
Here, the template parameters are:
Ris the type of reaction to advance with.Stateis the state vector that the KMC and reactions will advance.Tis the internal floating point or integer representation.
Tip
The KMCSolver C++ API is found at https://chombo-discharge.github.io/chombo-discharge/doxygen/html/classKMCSolver.html.
KMCSolver is designed to operate with the possibility of separating the solver from the reaction and state types.
Several template constraints exist on the reaction type R as well as the state type State.
State
The State representation must have a member function
/**
@brief Check if state is a valid state.
@return Returns false if any populations are negative.
*/
inline bool
isValidState() const noexcept;
This function should return true if the state is a valid one (e.g., no negative populations) and false otherwise. The functionality is used when using the hybrid advancement algorithm, see Hybrid algorithm.
Reaction(s)
The reaction representation R must have the following member functions:
/**
@brief Compute the propensity function for this reaction type.
@param[in] a_state State vector.
@return Reaction propensity.
@note User should set the rate before calling this routine.
*/
inline Real
propensity(const State& a_state) const noexcept;
/**
@brief Compute the number of times the reaction can fire before exhausting one of the reactants.
@param[in] a_state State vector.
@return Critical number of reactions.
*/
inline T
computeCriticalNumberOfReactions(const State& a_state) const noexcept;
/**
@brief Get the reactants involved in the reaction.
@return m_reactants
*/
inline std::list<size_t>
getReactants() const noexcept;
/**
@brief Get the state change due to a change in the input reactant species.
@param[in] a_particleReactant Reactant species.
@return State change for the reactant.
*/
inline T
getStateChange(const size_t a_particleReactant) const noexcept;
/**
@brief Advance the incoming state with the number of reactions.
@param[in,out] a_state State vector.
@param[in] a_numReactions Number of reactions.
*/
inline void
advanceState(State& a_state, const T& a_numReactions) const noexcept;
These template requirements exist so that users can define their states independent of their reactions.
Likewise, reactions can be defined to operate flexibly on state, and the KMCSolver can be defined without deep restrictions on the states and reactions that are used.
Defining states
State representations State can be defined quite simply (e.g. just a list of indices).
In the absolute simplest case a state can be defined by maintaining a list of populations like below:
class MyState {
public:
MyState(const size_t numSpecies) {
m_populations.resize(numSpecies);
}
bool isValidState() const {
return true;
}
std::vector<long long> m_populations;
};
More advanced examples can distinguish between different modes of populations, e.g. between species that can only appear on the left/right hand side of the reactions.
Defining reactions
See Implementation for template requirements on state-advancing reactions.
Using MyState above as an example, a minimal reaction that can advance \(A\rightarrow B\) with a rate of \(k=1\) is
class MyStateReaction {
public:
// List of reactants and products
MyStateReaction(const size_t a_A, const size_t a_B) {
m_A = a_A;
m_B = a_B;
}
// Compute propensity
Real propensity(const State& a_state) {
return a_state[m_A];
}
// Never consider these reactions to be "critical"
long long computeCriticalNumberOfReactions(const Mystate& a_state) {
return std::numeric_limits<long long>::max();
}
// Get a vector/list/deque etc. of the reactant's. <some_container> can be e.g. std::vector<size_t>
std::list<size_t> R::getReactants() const {
return std::list<size_t>{m_A};
}
// Get population
long long population(const size_t& a_reactant, const MyState& a_state) {
return a_state.m_populations[a_reactant];
}
// Advance state with reaction A -> B
void advanceState(const MyState& s, const long long& numReactions) const {
s.populations[m_A] -= numReactions;
s.populations[m_B] += numReactions;
}
protected:
size_t m_A;
size_t m_B;
};
Advancement routines
Many advancement routines for the KMCSolver are defined internally.
The most general one that uses the hybrid advance is
/**
@brief Advance using Cao et. al. hybrid algorithm over the input time. This can end up using substepping.
@param[in,out] a_state State vector to advance.
@param[in] a_dt Time increment.
@param[in] a_leapPropagator Which leap propagator to use.
@note Calls the other version with m_reactions.
*/
inline void
advanceHybrid(State& a_state,
Real a_dt,
const KMCLeapPropagator& a_leapPropagator = KMCLeapPropagator::ExplicitEuler) const noexcept;
When using the hybrid algorithm, the user should set the hybrid solver parameters through the function
/**
@brief Set solver parameters.
@param[in] a_numCrit Determines critical reactions. This is the number of reactions that need to fire before depleting a reactant.
@param[in] a_numSSA Maximum number of SSA steps to run when switching from tau-leaping to SSA (hybrid algorithm only).
@param[in] a_maxIter Maximum permitted number of iterations for the implicit solver.
@param[in] a_eps Maximum permitted change in propensities when performing tau-leaping for non-critical reactions.
@param[in] a_SSAlim Threshold for switching from tau-leaping of non-critical reactions to SSA for all reactions (hybrid algorithm only).
@param[in] a_exitTol Exit tolerance for implicit solvers.
*/
inline void
setSolverParameters(T a_numCrit, T a_numSSA, T a_maxIter, Real a_eps, Real a_SSAlim, Real a_exitTol) noexcept;
State and reaction examples
chombo-discharge maintains some states and reaction methods that can be useful when solving problems with KMCSolver.
The following two implementations are currently in use:
KMCSingleState, see the KMCSingleState C++ API and the KMCSingleStateReaction C++ API.KMCDualState, see the KMCDualState C++ API and the KMCDualStateReaction C++ API.
Verification
Verification tests for KMCSolver are given in
$DISCHARGE_HOME/Exec/Convergence/KineticMonteCarlo/C1$DISCHARGE_HOME/Exec/Convergence/KineticMonteCarlo/C2
C1: Avalanche model
An electron avalanche model is given in $DISCHARGE_HOME/Exec/Convergence/KineticMonteCarlo/C1.
The problem solves for a reaction network
In the limit \(X\gg 1\) the exact solution is
Figure Fig. 23 shows the Kinetic Monte Carlo solution for \(k_i = 2k_a = 2\) and \(X(0) = 10\).
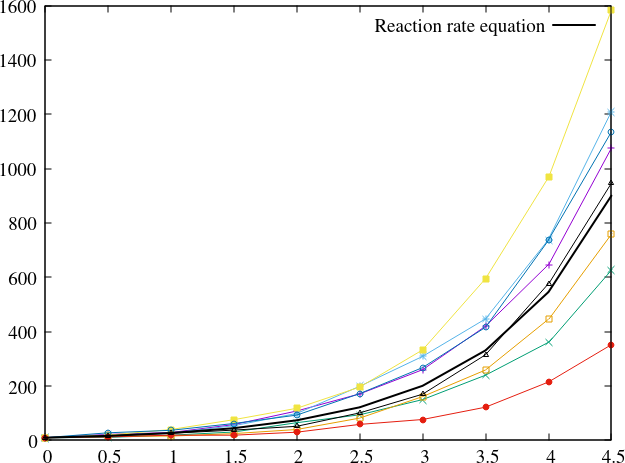
Fig. 23 Comparison of Kinetic Monte Carlo solution with reaction rate equation for an avalanche-like problem.
C2: Schlögl model
Solution the Schlögl model are given in $DISCHARGE_HOME/Exec/Convergence/KineticMonteCarlo/C2.
For the Schlögl model we solve for a single population \(X\) with the reactions
The states \(B_1\) and \(B_2\) are buffered states with populations that do not change during the reactions. Figure Fig. 23 shows the Kinetic Monte Carlo solutions for rates
and \(B_1 = 10^5\), \(B_2 = 2\times 10^5\). The initial state is \(X(0) = 250\).
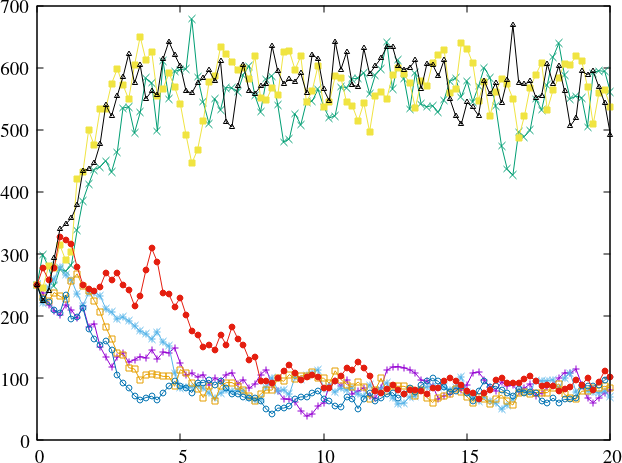
Fig. 24 Convergence to bi-stable states for the Schlögl model.